tags: NGS duplication
不管從哪個角度看我們都希望測序儀產出的數據中 duplicate 率盡量低。怎樣降低 duplicate 率? 構建文庫時,核酸提取質量要好,起始 DNA 量要足夠多,建庫過程中 PCR 循環數盡量少,可以的話構建 PCR-free 文庫最好,防范于未然。
什么樣的數據算是 duplicate
duplicate 就是一段序列的多個拷貝,以 PE 測序為例,用比對軟件在將測序 reads 比對到參考基因組之后,如果有兩對 reads 的 read1 和 read2 都完全比對到參考基因組上的相同位置,其中一對 reads 會被標記為 duplicate 。我畫了一個示意圖:
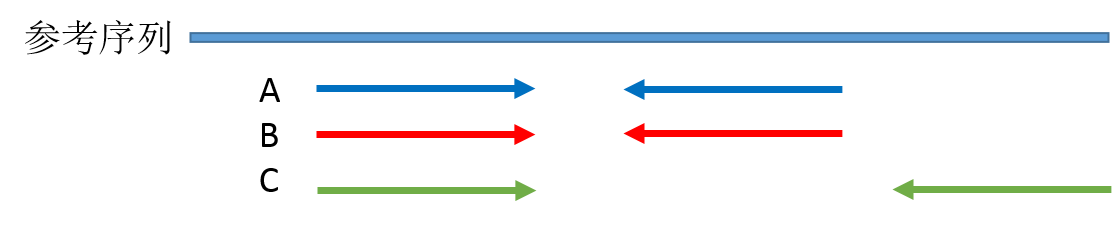
圖中 A 和 B 這兩對 reads 就是互相重復了,因為他們序列完全相同,這里說明一下,理論上 A 和 B 片段雖然兩端被測出來的序列完全相同,中間沒有被測到的堿基我們并不知道其序列是否也一樣,可能相同也可能不同,我們不得而知,但是現在我們只拿到了文庫片段 A/B 兩端的序列,所以只能根據現有的信息判斷 A/B 就是重復的,這也是 NGS 測序讀長短的弊端之一。片段 C 雖然其中一向序列與 A B 重復,但是 C 片段文庫片段比 A/B 長,另外一向的序列與 A/B 不同,因此不算 duplicate。
為什么會有 duplicate
要弄清楚這個問題,需要從 NGS 數據產出流程說起:
- 基因組核酸提取
- 基因組 DNA 隨機打斷,最常用的是超聲打斷。
- 被打斷的 DNA 片段經歷末端修復,3' 加A,兩端加接頭,選擇特定大小片段文庫進行 PCR 擴增(通過 PCR 擴增選擇性提高加上了接頭的文庫分子數量)。
- 文庫上機與 flowcell 上引物結合,經歷橋式 PCR 擴增形成 cluster 。
- 進行 SBS 測序,光學信號捕獲,生成序列。
我們首先假設基因組核酸提取是完整的基因組,打斷是完全隨機的(通常是這樣的)。
在第 3 步,PCR 擴增時同一個文庫分子會產生多個相同的拷貝,這是 duplicate 的主要來源(PCR duplicate)。
第 4 步,文庫中 DNA 片段與 flowcell 上引物結合,來源于同一個 DNA 片段的多個拷貝都結合到 flowcell 上,這樣會導致生成多個相同的 cluster,測序時也就有多個相同的序列被測出來,這些相同的序列就是 duplicate。
同在第 4 步,生成 cluster 時候一個 cluster 中的 DNA 鏈可能搭到旁邊另外一個 cluster 生成位點上,又長成一個相同的 cluster ,這也是 duplicate 的一個來源(Hiseq4000之后的 flowcell 會有的 cluster duplicate)。
第 5 步,一個 cluster 測序時的捕獲的熒光亮點由于形狀奇特,可能被軟件當成兩個熒光點來處理,這也產生了兩條完全相同的 reads。這個過程中可能產生完全相同的 reads。(光學 duplicate)
由此我們知道,PCR duplicate 特點是隨機分布于 flowcell 表面,光學 duplicate 特點是它們都來自 flowcell 上位置相鄰的 cluster 。cluster 的位置被記錄在 Fastq 文件 @seq-id 這一行中。
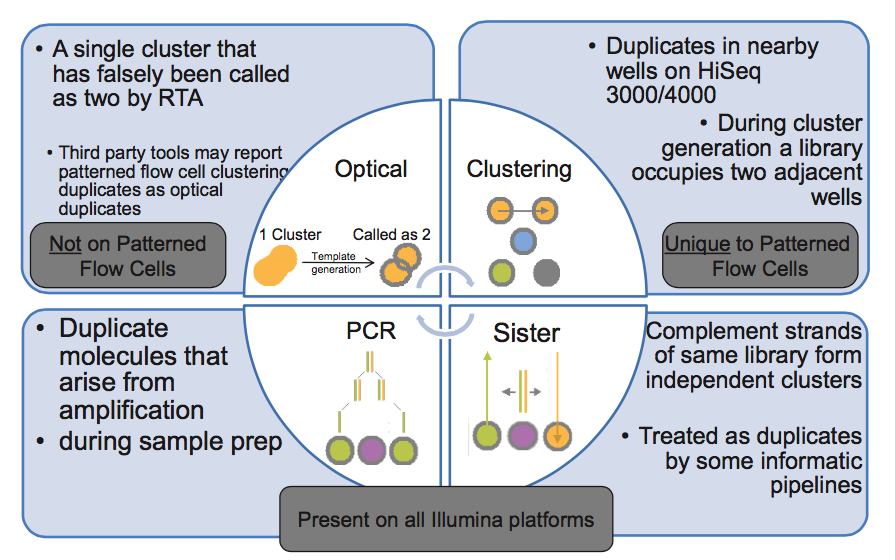
下圖的右下角還有一種 duplicate 來源,sister? 這種一個文庫分子的兩條互補鏈同時都與 flowcell 上的引物結合分別形成了各自的 cluster,最后產生的兩對 reads 完全反向互補,map 到參考基因組也分別在正負鏈上的相同位置,有的分析中也算 duplicate,雖然我遇到的這種正負鏈測序結果通常是不算 duplicate 的。
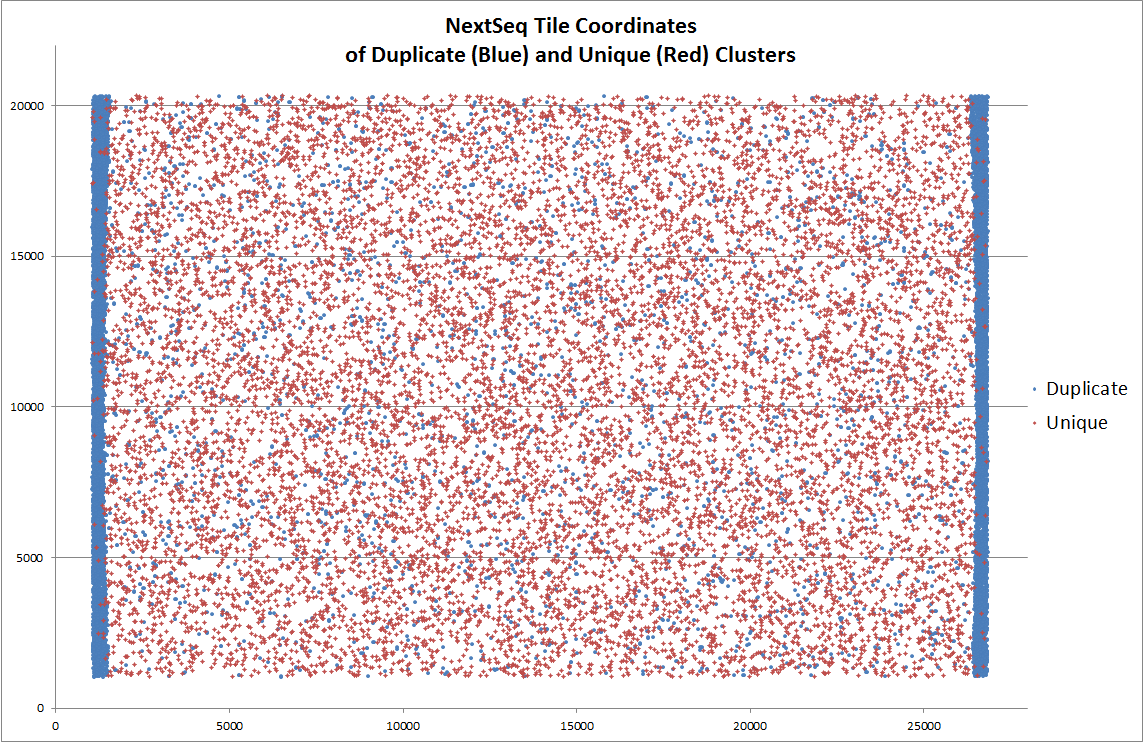
另外,據說 NextSeq 平臺上出現過由于熒光信號捕獲相機移動位置不夠,導致 tile 邊緣被重復拍攝,每次采樣區域的邊緣由于重復采樣而出現的 duplicate,下圖中藍色點代表 duplicate ,在 tile 兩側明顯富集。Illumina 公司回應說這沒毛病,符合預期……
總結一下,duplicate 的產生即有可能來自實驗過程,也有可能來自測序儀。
PCR 將模板擴增了數千倍,但數據中 duplication 率只有 15%
我曾經有這樣的疑惑,為什么文庫構建過程中的 PCR 將每個文庫分子都擴增了上千倍,以 PCR 10個循環為例 2^10= 1024 ,但是實際測序數據中 duplication 率并不高(低于20%)。后來我看到一篇文章從統計概率的角度詳細探討了一下 duplication 率的影響因素,順便一提,這個博主的故事也很令人佩服。
PCR 的過程中不同長度的文庫分子被擴增的效率不同(GC 太高或 AT 含量太高都會影響擴增效率),PCR 更傾向于擴增短片段的文庫分子,這里先不考慮文庫片段擴增效率的差異,把問題簡化一下,假設所有文庫分子擴增效率都相同。PCR duplicate 的主要來源是同一個文庫分子的不同拷貝都在 flowcell 上生成了可以被測序的 cluster ,導致同一個分子的序列被測序儀讀取多次。那么為何在每個分子都有上千個拷貝的情況下,實際卻很少出現同一分子的多個拷貝被測序的情況呢?主要原因就是文庫中 unique 分子的數量比被 flowcell 上引物捕獲的分子數量多很多,直白點說就是 flowcell 上用于捕獲文庫分子的引物數量太少了,兩者不在同一個數量級,導致很少出現同一個文庫分子的多個拷貝被 flowcell 上引物捕獲生成 cluster。
假設文庫中所有分子與引物的結合都是隨機的,簡化一下就相當于,一個箱子中有 n 種顏色的球(文庫中的 n 種 unique 分子),每種顏色有 1000 個(PCR 擴增的,隨 cycle 數變化),從這個箱子中隨機拿出來 k 個球(最終測序得到 k 條 reads),其中出現相同顏色的球就是 duplicate,那么 duplication 率就可以根據有多少種顏色的球被取出 0,1,2,3…… 次的概率計算,可以近似用泊松分布模型來描述。
以人全基因組重測序 30X 為例,PE150 需要約 3x10^8條 reads ,文庫中 unique 分子數其實可以通過上機文庫的濃度和體積(外加 PCR 循環數)計算出來,這里用近似值 3.5x10^10 個 unique 分子。每個 unique 分子期望被測序的次數是 3x108/3.5x1010 = 0.0085 ,每個 unique 分子被測 0,1,2,3… 次的概率如下圖:
> x <- seq(0,10,1)
> xnames <- as.character(x)
> xlab <- "一個文庫分子的所有拷貝被測序的次數"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.0085),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
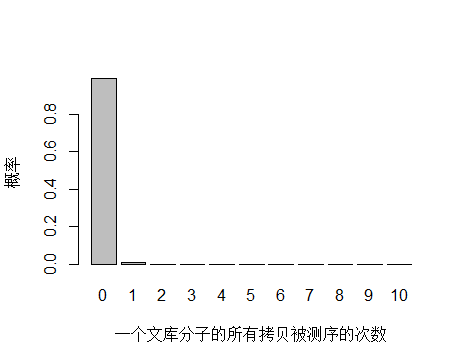
由于 unique 分子數量太多,被測 0 次的概率遠高于 1 和 2 次,我們去除 0 次的看一下:
> x <- seq(1,10,1)
> xnames <- as.character(x)
> xlab <- "一個文庫分子的所有拷貝被測序的次數"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.0085),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
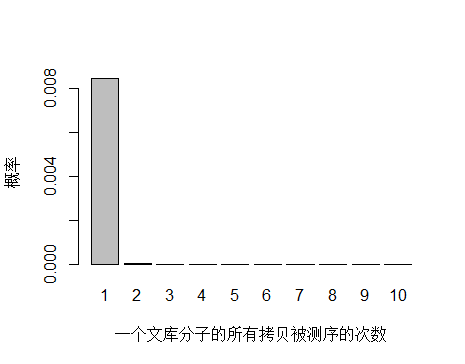
unique 分子被測序 1 次的概率遠大于 2次及以上,即便一個 unique 分子被測序 2 次,我們去除 duplicate 時候還會保留其中一條 reads。
如果降低文庫中 unique 分子數量到 4.5x10^9 個,PCR 循環數增加以便濃度達到跟上面模擬的情況相同,測序 reads 數還是 3x10^8 條,每個 unique 分子預期被測序的次數是 3x108/4.5x109 = 0.067 。
> x <- seq(1,10,1)
> xnames <- as.character(x)
> xlab <- "一個文庫分子的所有拷貝被測序的次數"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.067),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
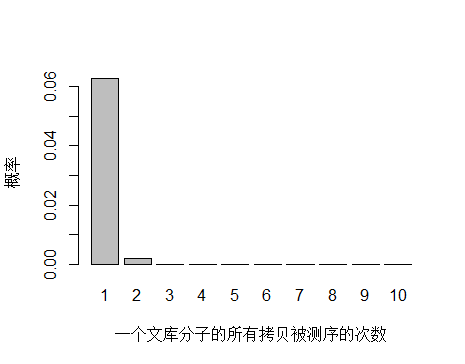
unique 分子數量減少,被測序 2次的概率增大,duplication 率顯然也會增高。
到這里已經可以很明白的看出 duplication 率主要與文庫中 unique 分子數量有關,所以建庫過程中最大化 unique 分子數是降低 duplication 率的關鍵。文庫中 unique 分子數越多,說明建庫起始量越高,需要 PCR 的循環數越少,而文庫中 unique 分子數越少,說明建庫起始量越低,需要 PCR 的循環數越多,因此提高建庫起始量是關鍵。
